Urgent Info
- There is no Urgent Info now.




GSIC
| Addr. | 2-12-1 O-okayama, Meguroku, Tokyo 152-8550 JAPAN |
| Contact this mail address. |
You are here
Development of an ultra-fast computing pipeline for metagenome analysis with next-generation DNA sequencers
Akiyama Laboratory, Department of Computer Science, Tokyo Institute of Technology
info[at]bi.cs.titech.ac.jp
Background
Metagenome analysis is the study of the genomes of uncultured microbes obtained directly from microbial communities in their natural habitats. The analysis is useful for not only understanding symbiotic systems but also watching environment pollutions. However, metagenome analysis requires comparisons of sequence data obtained from a sequencer with sequence data of remote homologues in databases because current databases do not include sequence data for most of microbes in the sample. For that purpose, mapping software used in the genome analysis performed for known organisms are insufficient because comparison between sequences of remote homologues has to consider mutations, insertion and deletion. Therefore, sensitive sequence homology search processes are required in metagenome analysis. Unfortunately, this process needs large computation time and is thus a bottleneck in current metagenome analysis based on the data from the latest DNA sequencers generally called next-generation sequencers.
Methods
We developed a fully automated pipeline for metagenome analysis that can deal with huge data obtained from a next generation sequencer in realistic time by using the large computing power of TSUBAME 2.0 supercomputer.
In the pipeline, two different sequence homology search tools can be selected;
1) BLASTX, standard sequence homology search software used in many metagenomic researches
2) GHOSTM, GPU-based fast sequence homology search software
GHOSTM is our original sequence homology search program. The program is implemented by using NVIDIA's CUDA and able to search homologues in a short time by using GPU-computing. The program with high sensitivity setting (the length of indexes in the initial search process as K=3) shows high search sensitivity which is comparable with BLAST and even the program with high speed setting (the length as K=4) have much higher sensitivity than BLAT, a well-known fast homology search program, and its sensitivity is enough for metagenome analysis (Fig. 1).
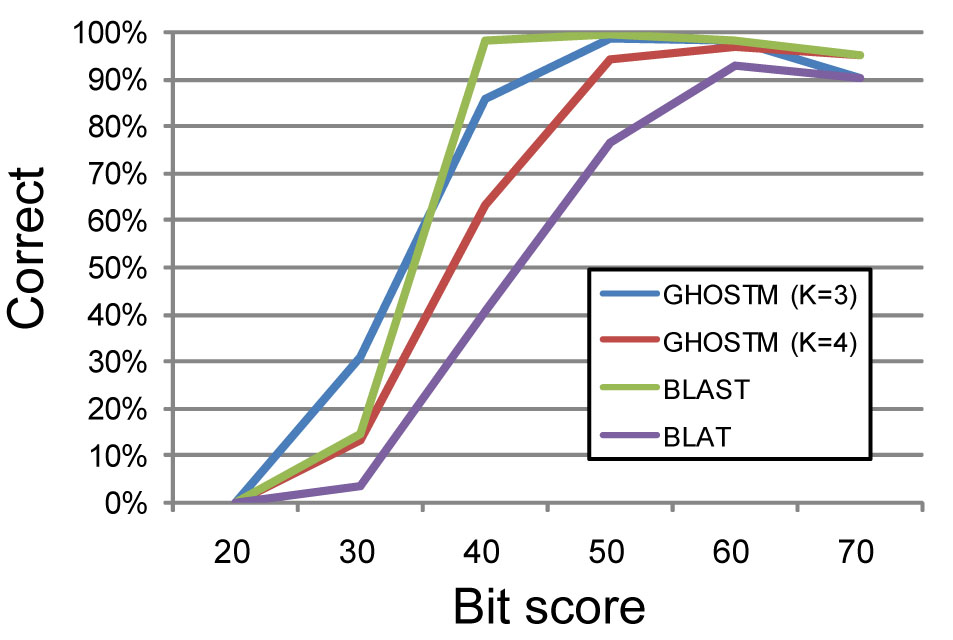
Fig.1. comparison of sensitivities of homology search programs
Experiments
We performed a large-scale metagenome analysis by using our pipeline. We used data sampled from polluted soils and obtained by using a next-generation sequencer. We evaluated the effective performance of the pipeline with both 1) BLASTX on CPUs and 2) GHOSTM on GPUs.
Original metagenomic data: 224 million DNA reads (75 bp)
Size after excluding low-quality data: 71 million DNA reads
Homology search: 71 million DNA reads vs. NCBI nr amino-acid sequence DB (4.2GB)
Results
File I/O processes including a database copy and writing search results caused a contention problem when we used many computation nodes. Thus, we changed to employ a sophisticated file transfer manner where data are simultaneously copied from local disk of a node to another in a binary-tree manner. As results, the pipeline shows almost linear speedup to the number of computing cores. When we use BLASTX as a homology search program, the pipeline achieves to process about 24 million reads per an hour with 16,008 CPU cores (1,334 computing nodes) (Fig. 2). When we use GHOSTM (K=4) as a homology search program, the pipeline achieves to process about 60 million reads per an hour with 2,520 GPUs (840 computing nodes) (Fig. 3).
These results indicate the pipeline can process genome information obtained from a single run of next generation sequencers in a few hours. We believe our pipeline will accelerate metagenome analysis with next generation sequencers.
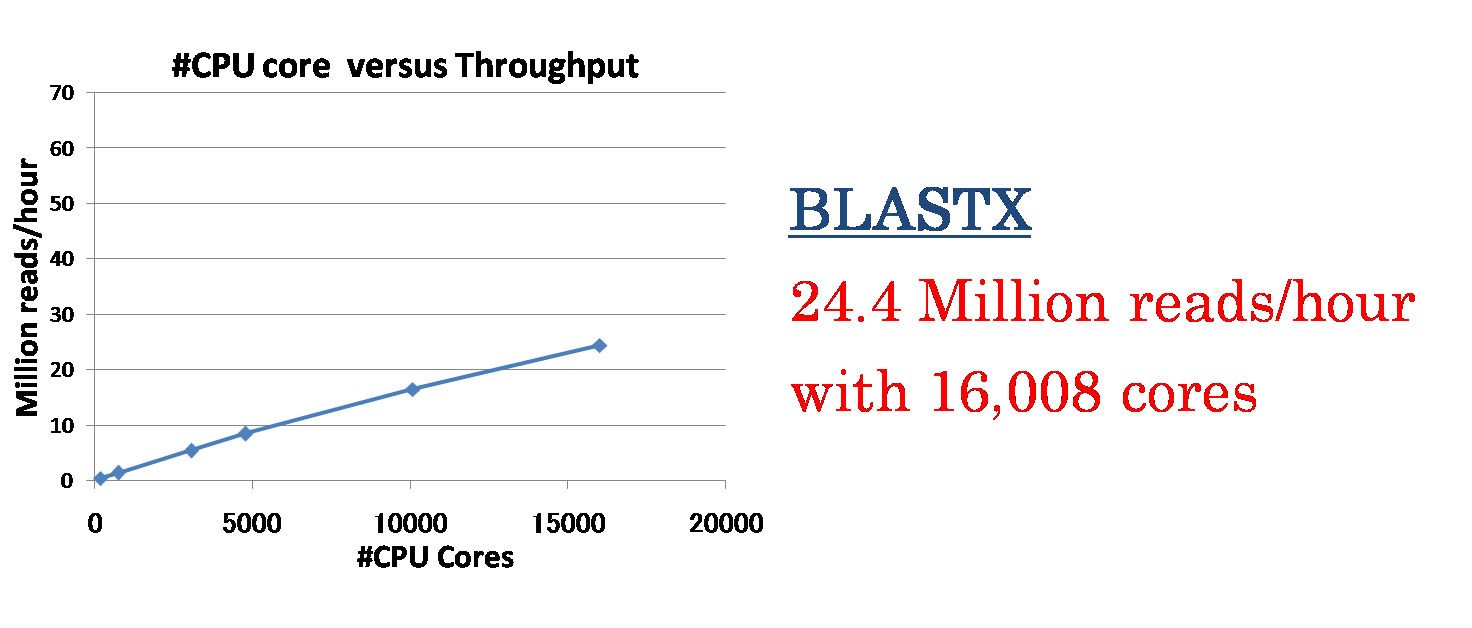
Fig. 2. Speedup of the BLASTX-based system for the number of CPU cores
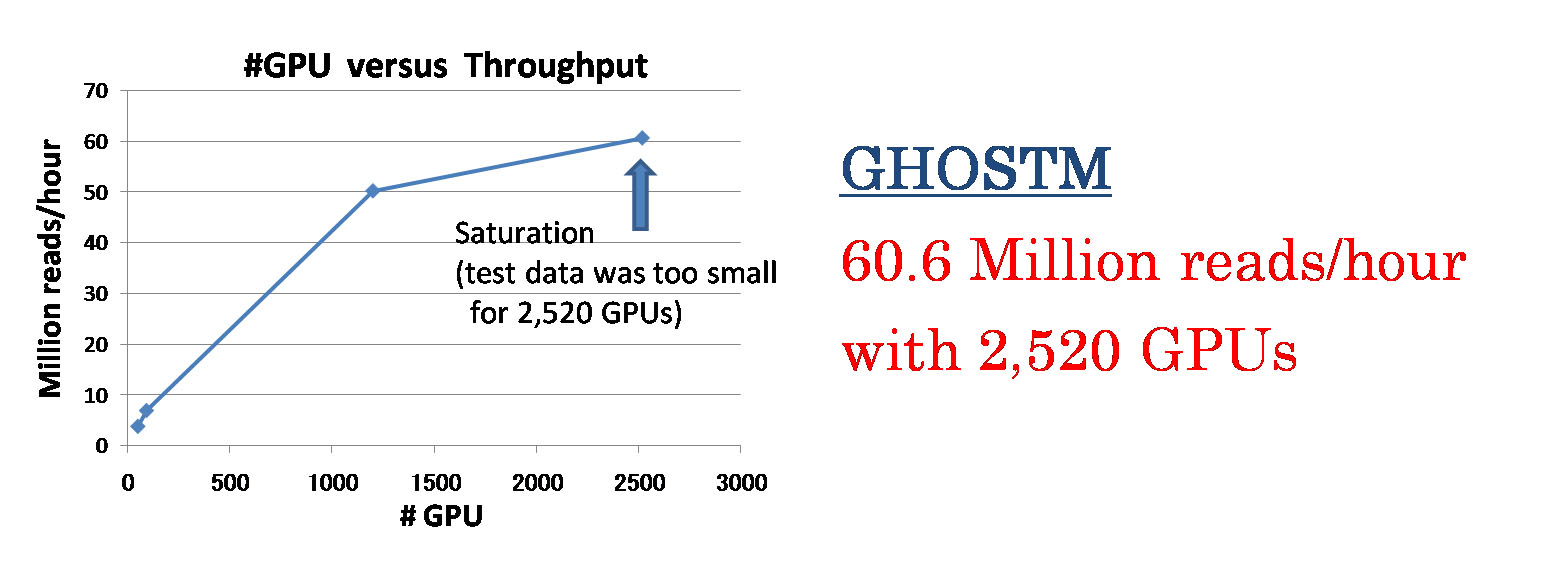
Fig. 3. Speedup of the GHOSTM-based system for the number of GPUs