



You are here
次世代シーケンサーを用いたメタゲノム解析向けの超高速パイプラインの構築
東京工業大学 大学院情報理工学研究科 計算工学専攻 秋山研究室
info[at]bi.cs.titech.ac.jp
背景・目的
土壌等の環境中に生息する微生物のゲノムを分離培養せずにそのまま丸ごとシークエンスして解析するメタゲノム解析は、未知の微生物に関してのゲノム情報が得られるだけでなく、その環境中の共生系の理解や環境汚染の監視等に有用です。しかし、メタゲノム解析では、サンプルに含まれる多くの種のゲノム情報がデータベースに登録されていないため、遠縁の種の配列データとの間で比較が必要となります。それには既知の生物に対するゲノム解析で行われるような完全一致に基づく検索では不十分であり、置換や挿入、欠失を考慮した配列相同性検索処理が必要となります。この処理には膨大な計算が必要であり、そのため、この相同性検索に要する計算時間が次世代シークエンサーと呼ばれる新型のシークエンサーによるメタゲノム解析を進める上でのボトルネックの一つとなっています。
方法
本研究ではTSUBAME2.0の膨大な計算能力を利用し、次世代シークエンサによる大量のメタゲノム情報を現実的な時間内に解析可能とする大規模な全自動解析パイプラインを構築しました。
また、そのパイプライン中で行われる配列相同性検索には以下の2つのプログラムを利用可能としました。
1)従来のメタゲノム研究で標準的に利用されてきた配列相同性検索プログラムBLASTX
2)BLASTXと同等の検索感度を持つオリジナルのGPUプログラムGHOSTM
GHOSTMは我々が独自に開発した配列相同性検索プログラムであり、NVIDIA社のCUDAを用いて実装されており、GPU計算によって高速な処理を可能としています。高検索感度設定(初期探索時のインデックスの長さK=3)の際にはBLASTとほぼ同等の検索感度を持ち、また、低検索感度設定(K=4)の際でもメタゲノム解析に十分な検索感度を保ち、高速な相同性検索プログラムとして知られるBLATに比べ高い検索感度を持っています(図1)。
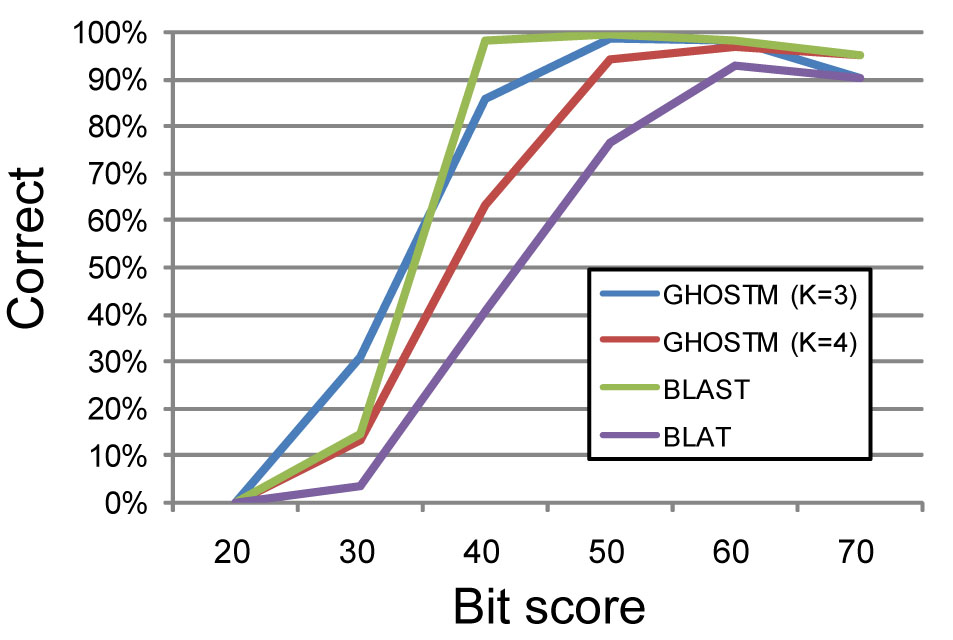
図1.相同性検索プログラムの検索感度の比較
実施内容
下記に示す、次世代シークエンサによって得られた汚染土壌に関するメタゲノムデータに対し、作成されたパイプラインを用いた大規模な解析を行い、1)CPU上で動作するBLASTXと2)GPU上で動作するGHOSTMのそれぞれについて実効性能を比較しました。
メタゲノム原データ: 224 million DNAリード (75 bp)
低品質データ除去後: 71 million DNAリード
相同性検索: 71 million DNAリード vs. NCBI nr アミノ酸配列DB (4.2GB)
結果
多くの計算ノードを利用する大規模な計算で問題となる、データベースのコピー、計算結果の書き込みといったファイルの入出力に関する処理について、ノード間でコピーを2分木状に行う工夫を行う事で、パイプラインは計算コア数に対してほぼ線形の速度向上を示しました。また、1)BLASTXを相同性検索に用いた場合、TSUBAME2のCPU 16,008コア(1,334ノード)用いた際に1時間あたり約24 millionのDNAリードの処理を実現し、2)GHOSTM(K=4)では2,520 GPU(840ノード)を用いることで1時間あたり約60 millionのDNA リードの処理を実現しました。
これらの結果は次世代シークエンサーの一度の読み取り実行によって得られるゲノム情報を数時間の内に処理可能な事を示しており、今後我々のパイプラインによって次世代シークエンサーによるメタゲノム解析が促進されると考えられます。
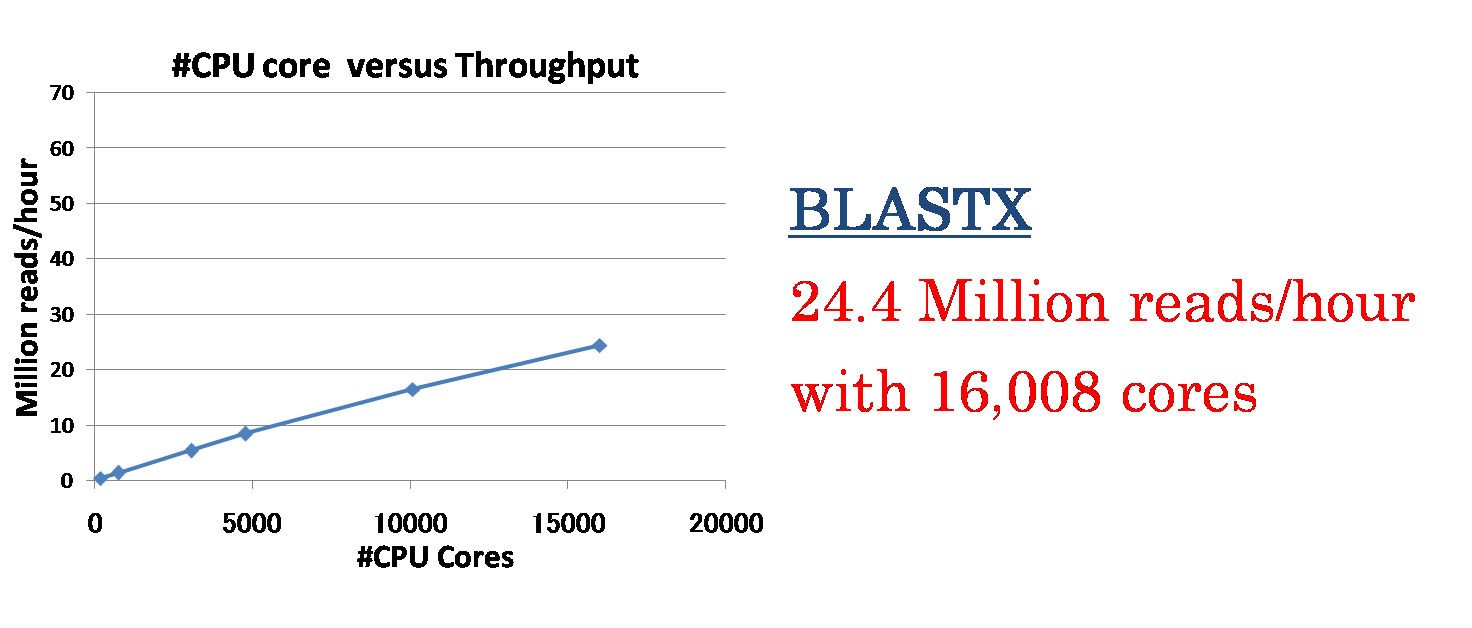
図2.BLASTXを用いた際のCPUコア数に対する処理速度の向上
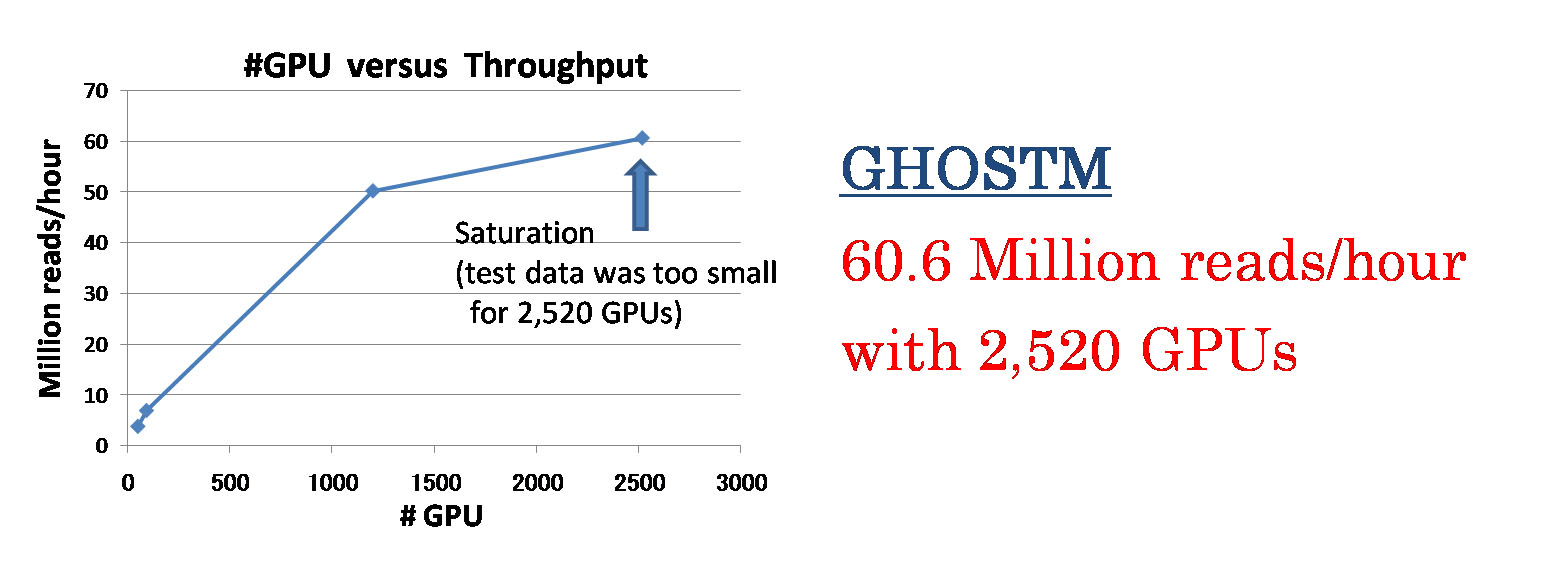
図3.GHOSTMを用いた際のGPU枚数に対する処理速度の向上