



You are here
Molecular Dynamics Simulation of a Biomolecule with High Speed, Low Power and Accuracy using GPGPU
Sekijima Laboratory
sekijima [at] gsic.titech.ac.jp
http://www.bio.gsic.titech.ac.jp
Background
Molecular Dynamics (MD) simulation is a powerful tool for simulating the motion of molecules and obtaining detailed information on the interesting systems and phenomena. During the simulation, we iteratively integrate numerically the equation of motion of the whole system and the time series of the positions and momentum of every molecule or atom, which is called the trajectory results from the simulation. For this reason, MD is widely used in computational biology especially for studying the motion of proteins. In our research, we have showed the usefulness of GPU-accelerated MD simulation from three viewpoints. One is the speed of computation, another is the energy consumption and the other is the accuracy.
Methodology
- Nucleosome (25095 Atoms)
- PMEMD -SPDP mode for GPU MD from AMBER11
- Implicit Solvent(GB) model
- NVE ensemble

Result
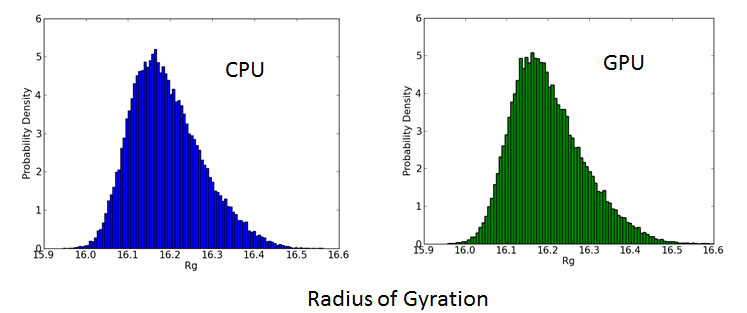
In this study, we performed molecular dynamics simulation using GPU on TSUBAME2.0 supercomputer. Then the performance performance and result was compared to those of conventional CPU molecular dynamics regarding calculation speed, energy consumption and accuracy. Our experiment showed that using GPUs could yield a great improvement in both the calculation speed and energy consumption. Comparing 8 nodes 16 GPUs result and 8 nodes 96 CPU cores result, we could achieve about 10 times acceleration and 75 % energy reduction. And the results of GPU molecular dynamics was consistent with
that of CPU molecular dynamics within acceptable range.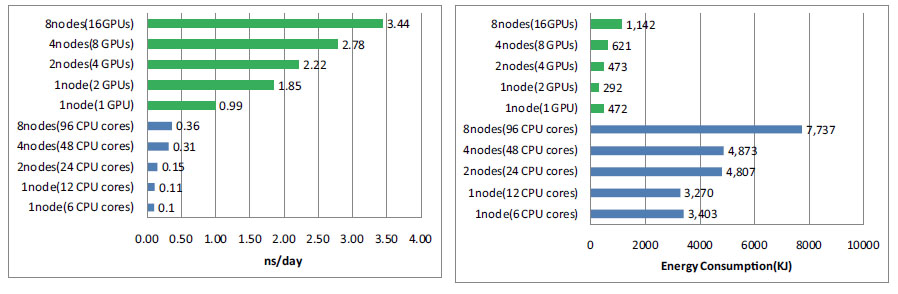
Reference
Shiqiao Du, Takuro Udagawa, Toshio Endo and Masakazu Sekijima"Molecular Dynamics Simulation of a Biomolecule with High Speed, Low Power and Accuracy Using GPU-Accelerated TSUBAME2.0 Supercomputer", APSIPA ASC 2011, accepted